不整脈科
対象疾患・治療法
先天性QT延長症候群
先天性QT延長症候群
QT延長症候群(LQTS)は、心電図上のQT時間延長とT波の形態変化を特徴とし、ときにTorsade de Pointes(TdP)と称されるQRSの極性と振幅が心拍ごとに刻々と変化する多形性心室頻拍を認め、失神や心臓突然死の原因となる症候群である(図1)。
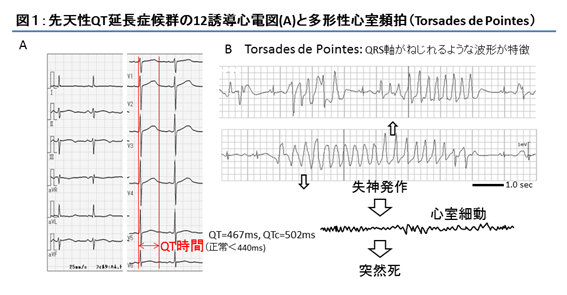
先天性LQTSは、無症状で学校・職場検診の心電図検査で指摘される場合や、失神発作や心停止を契機に診断される場合が多く、その頻度は6歳児で約3000人に1人、12歳児では1000人に1人と推定される1)。先天性LQTSは遺伝学的検査が保険適応となっており、遺伝学的検査が診断のみならず治療や予後予測などにも重要な役割をなしている。
1. QT延長症候群の診断
先天性LQTSの診断は、心電図が重要である。繰り返し記録された12誘導心電図でBazett補正後のQTcが500ms以上であれば診断確実である。あるいは心電図、症状、家族歴などを点数化したLQTSリスクスコア(Schwartzスコア:表1) が3.5点以上あればLQTSと診断される。さらに病的変異を認めずQT延長をきたす2次性因子がない状況で、QTc:480~499msを示しかつ説明のつかない失神を認める場合もLQTSと診断可能である2)。
表1.LQTSリスクスコア(Schwartzスコア)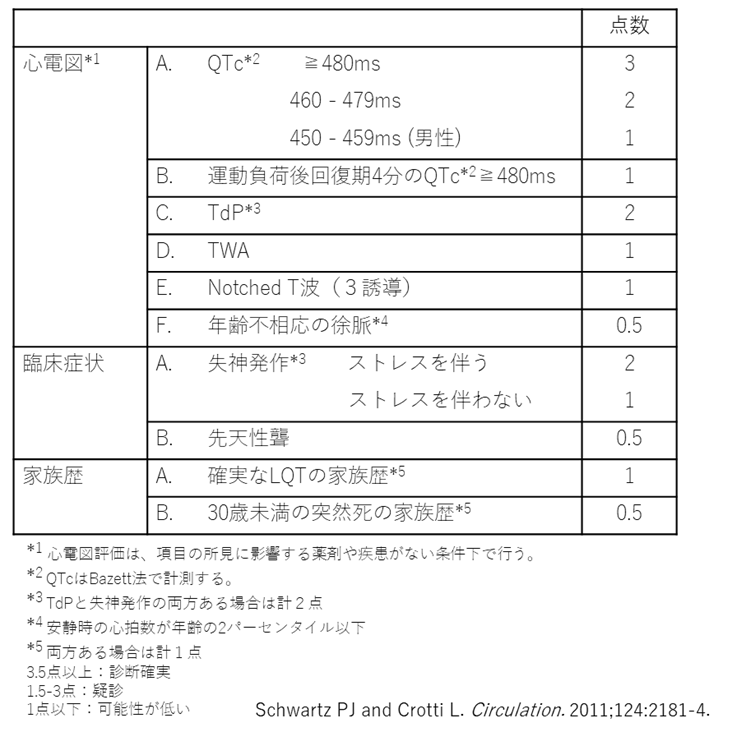
先天性LQTSは常染色体顕性遺伝のRomano-Ward症候群(RWS)と、常染色体潜性遺伝で難聴を伴うJurvell and Lange-Nielsen症候群(JLN)に分けられてきた。頻度は前者が圧倒的に多く、さらに遺伝子診断が一般的になった昨今では「遺伝型」で分類される。したがって、先天性LQTSの診断でも上記の「表現型」(QT延長や不整脈の有無)とは関係なく、LQTS関連遺伝子(表2)に病的バリアント(変異)を認める場合にも診断される。LQTSの原因として過去17の遺伝子が報告されたが、ClinGenにより再評価され最終的には表2の遺伝子がLQTSの原因とされた3)。さらにその中で実際同定される遺伝子のほとんど(90%)がKCNQ1 (LQT1), KCNH2 (LQT2), SCN5A (LQT3)である。また後天性(2次性あるいは薬剤性)LQTSでも3割近くが遺伝的背景を有する。
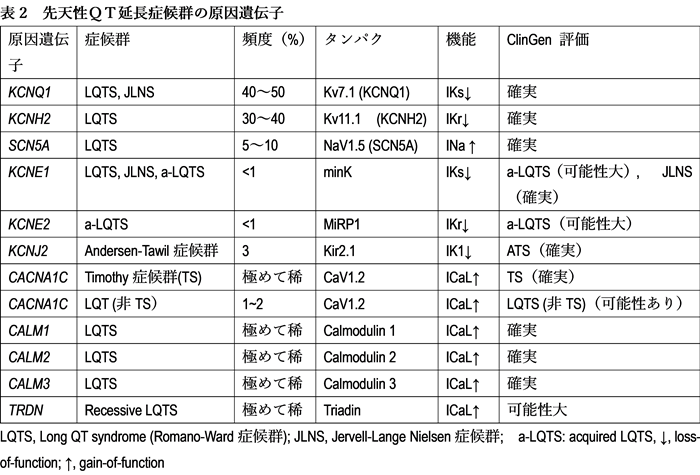
2. 遺伝学的検査の適応
LQTSに対する遺伝学的検査は、臨床経過、家族歴、心電図からLQTSと診断あるいは循環器医が強く疑う場合は、LQTSの原因として確実な遺伝子に対する遺伝学的検査を行うことが推奨(クラスI)されている。また発端者に同定された先天性LQTSの原因遺伝子変異に対する、家族または血縁者の遺伝学的検査も積極的に奨められる(表3)
これまではKCNQ1, KCNH2, SCN5AがLQTSの遺伝学的検査の主な対象遺伝子であったが、2024年ガイドラインではこれらに加えてCALM1~3や、KCNE1, CACNA1C, KCNJ2, TRDNなども疾患原因遺伝子として明記された。

3. イオンチャネルと不整脈発生機序
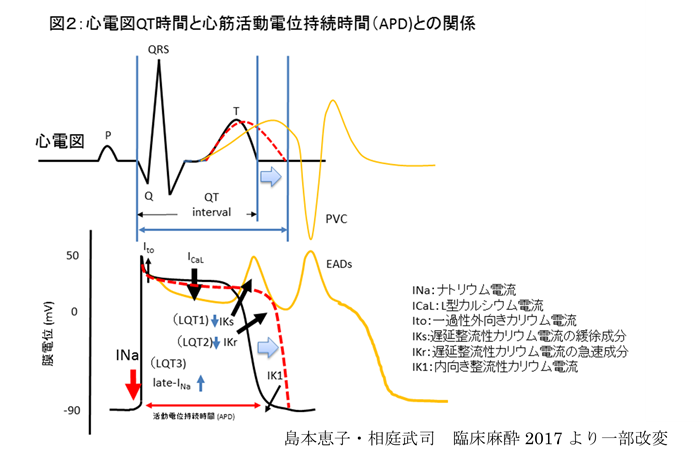
代表的な先天性LQTSでは、心室筋細胞の外向き電流(LQT1:IKs, LQT2: IKr)が減少(loss of function)するか、または内向き電流(LQT3: late INa)が増加(gain of function)することにより活動電位持続時間(APD)が延長し、心電図上のQT時間の延長を呈する。さらに活動電位のプラトー相付近の時間が長くなることから内向きCaチャネルが再活性化し、早期後脱分極(EAD)を生じると不整脈につながると考えられる(図2)。
4. 日本におけるLQTSのエビデンス
我々はLQTS研究班(厚労科研難治性疾患政策事業)の活動を通じ、2024年末までに計3851人のLQTS患者を全国19施設から登録した4)。LQTSの臨床的診断基準(QTc≧500msまたはSchwartsスコア≧3.5)を満たす例は2409人(62.5%)で、そのほとんどが遺伝型も陽性であった。一方、表現型は必ずしもLQTSの診断基準を満たさないが、遺伝型が陽性でLQTSと診断された例も1439人(37.3%)あり、その約6割は発端者の家族であった(図3A)。
登録患者全体でLQTSと診断された年齢(図3B)は10歳前後が最も多いが、家族では40歳台に診断された例も多く認めた。これは発端者(子供)が診断された結果、家族調査で両親などが診断されるケースが多いためと推定される。このようにLQTS患者は小児だけでなく、成人にも幅広く存在する。さらに診断時QT間隔(QTc)は、発端者では500ms前後に対して、家族の多くは心電図QT間隔が正常あるいは境界域である(図3C)。このように同じLQTSであっても発端者と家族の表現型には大きな違いがある。
登録患者の遺伝型の内訳(図2D)は、これまでと同様にKCNQ1, KCNH2, SCN5Aの順で多く、この主要3遺伝子にて約9割を占めていた。一方、これら以外の遺伝型についても一定数を認め、特にKCNJ2やCACNA1Cは両者で5%近くを占めていた。KCNJ2変異は短指症や小顎症、周期性四肢麻痺などの心外病変を合併することもありAndersen-Tawil症候群(ATS)と呼ばれ、典型的なQT延長ではないが心電図上U波と特徴的な2方向性心室期外収縮(PVC)を示し、特発性PVCやCPVTとの鑑別も必要である。CACNA1C変異(LQT8)の一部はTimothy症候群(TS)とも呼ばれるが、非TS型もあり、徐脈や房室ブロックを伴いLQT3似の遅発性T波を呈する。LQTSで通常のLQT1~3の遺伝子に病的バリアントを認めない場合には、これらの比較的頻度の少ない遺伝子についても検査する必要がある。
さらに最近注目されているCALM1~3はいずれもカルモジュリン蛋白をエンコードする遺伝子で、その変異は非常に稀で国内でもわずか13例を認めたに過ぎないが、10歳前後で半数以上が致死性不整脈を生じ他の遺伝型と比べても極めて重篤である4)。またカルモジュリンの機能異常はLQTSの他、CPVTや特発性心室細動にも関係している。

5. 遺伝型別の心事故の誘因と生活指導
頻度の多いLQT1~3では遺伝型に特異的な不整脈の誘因が知られている。図4のように、LQT1患者における失神発作、心停止、突然死などの心事故の多くは運動中に(特に持続する運動、マラソン、水泳)に多い。LQT2患者の心事故は、情動ストレス(恐怖や驚愕)、睡眠中の雑音(目覚まし時計など)による覚醒時など、急激に交感神経が緊張する場合が知られている。しかし最近実施したLQTS登録研究では、LQT2の発作の誘因は情動ストレスのみならず、むしろ安静・睡眠中(時間帯では朝方に多い)のイベントが多いことが判明した。LQT3患者も、心事故の多くは非運動時、夜間、睡眠中や安静時に多いため、発作の誘因のみでLQT2とLQT3を見分けることは容易ではない。
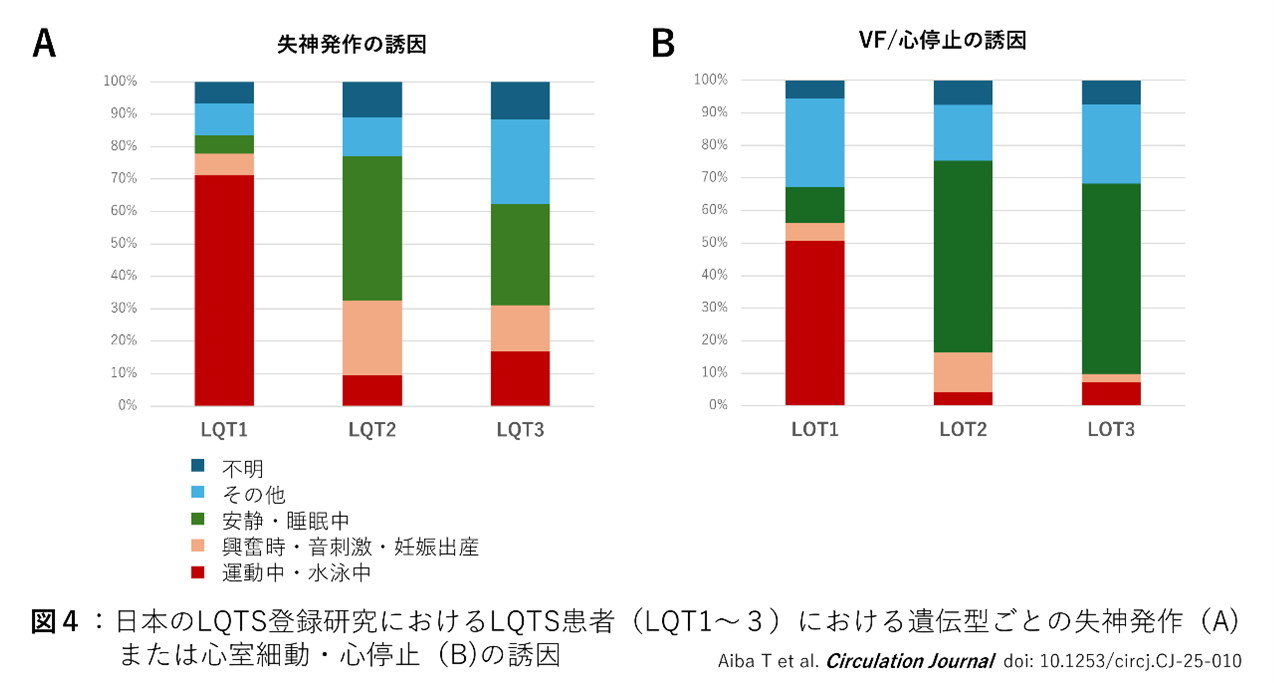
上記結果から、LQT1患者に対して運動制限は必須である。仮に無症候の場合でも、LQT1患者は少なくとも競争的スポーツは避けることが望ましい。LQT1では水泳中の心事故が特徴的であるため、特に未成年者では競泳、潜水などは禁止する必要がある。LQT1以外の運動制限については、必ずしもエビデンスはなく、多くの場合は学校生活管理区分表のE禁(水泳は監視下)が推奨される5)。またLQT2では思春期以降に女性でリスクが高く、また妊娠・出産後にQT延長が増悪し心イベントが発生することがあり、ハイリスク患者においてはβ遮断薬など治療を継続したうえでの妊娠・出産が望ましい。
なお運動参加に伴う心事故のリスクは、専門医による評価と適切な医療を受けて管理されたLQTS患者では必ずしも高くはないことが北米を中心とする研究グループから示され、運動制限を緩和する動きも広がりつつある。ただし我が国での学校検診を中心としたLQTS患者の診断・管理とは臨床背景などもやや異なっており、LQTS患者の競技スポーツへの参加をどこまで許容できるかについては今後議論が必要である。
6. 遺伝子検査と重症度・予後
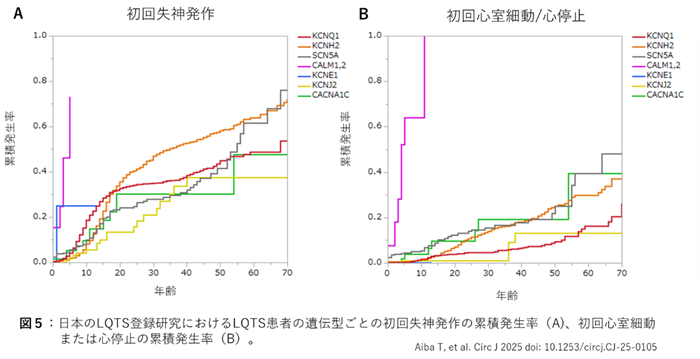
LQTSは遺伝型によって不整脈リスクが異なる。初回発作の累積発生率(図5)はCALM遺伝子例が極めて高くほぼ10歳までに致死性不整脈が発症する。CALM例を除けば、無治療の場合20 才までに約3割の患者で失神発作を経験する(図5A)。さらに成人以降はKCNH2(LQT2)の発作頻度が高い。心室細動(VF)/心停止に限るとKCNH2(LQT2)に加えてSCN5A(LQT3)やCACNA1C(LQT8/TS)もKCNQ1(LQT1)やKCNJ2(ATS)に比べて高リスクである(図5B)。このように遺伝型がわかることで、大まかな予後との関係が明らかとなる利点がある。さらにLQTSでは各遺伝型のバリアントの種類による重症度 や治療に対する反応性の違いも報告されている。
7. 遺伝型による治療方針
先天性LQTSでは遺伝型に即した治療が実践されている。LQT1ではβ遮断薬の有効性が最も高く、LQT2でも第一選択薬はβ遮断薬である。さらにβ遮断薬のなかでも、β1非選択性のナドロールまたはインデラルが推奨され、特にLQT2についてはナドロールが強く推奨される。先天性LQTSに対するβ遮断薬の適用は、失神やVT/VFなどがあれば治療開始すべきであるが、例え無症状の患者であってもQTc時間が470ms以上あれば、内服を推奨すべきと考えられる(表4)。また無症状でQTc<470msでも、性別や遺伝子型によってその適応が考慮されている。
表4 LQTSに対するβ遮断薬治療の適応
β遮断薬以外では、メキシレチン、ベラパミルなどが、β遮断薬との併用で補助的効果が期待される。メキシレチンはLQT3に対して有効であることが知られているが、LQT2に対してもβ遮断薬との併用により心イベントを抑制したエビデンスが報告されている。また低K血症はQT延長を助長するため、K +製剤やK+保持性利尿薬によって一定水準(>4.0mEq/l)の血清K+値を維持することが望ましい。
デバイス治療:心室細動・心停止の既往例や、十分な薬物治療にもかかわらず致死性不整脈再発を認める例では、いずれの遺伝型でも植込み型除細動器(ICD)の適応となる。一方、無症状で適切な薬物治療も試されていない症例に対してICD植え込みは行わない。ペースメーカ治療は、徐脈を合併または徐脈依存性にQT延長が顕在化する症例には有効である。
8. 国立循環器病研究センターにおけるLQTSの遺伝子検査
国立循環器病研究センター(国循)では遺伝性検査室において次世代シーケンサー(NGS)とSanger法を用いて遺伝子検査を行っており、LQTSに対しては2024年ガイドライン(表2)に基づき、KCNQ1, KCNH2, SCN5Aの他にCALM遺伝子(CALM1~3)、KCNE1, KNCJ2, CACNA1Cなどを保険診療として検査しています(遺伝子診断料:8000点、遺伝カウンセリング加算:1000点)。本検査は臨床検査のISO15189に準拠して品質精度管理を実施しています。また得られた遺伝子解析結果は、循環器医と遺伝子検査室、ゲノム医療部などの遺伝診療に携わるエキスパートパネルによってLQTSに関連するバリアントの解釈を実施しています。外部施設からの検査委託も受付しており、遠方の患者様については国循まで来院頂かなくても、近隣のご施設から検体を送ることで検査を受けることが可能です。検査結果は平均2か月(お急ぎの場合は1か月程度)で返却可能です。
LQTS関連遺伝子については、病的意義不明のバリアントも存在し必ずしも全てのバリアント情報がClinVarやACMGなどで病的か否かを判断できるとは限りません。我々は4000例以上のLQTSデータベースの登録情報をもとに、遺伝情報と臨床情報を総合的に判断し遺伝子検査の判定を行っています。国立循環器病研究センターでは、網羅的、迅速かつ正確にLQT関連の遺伝子バリアントを診断し、豊富な臨床データの蓄積をもとに遺伝型・バリアントごとの生活指導、治療等の助言をおこなっており、個別治療(Precision Medicine)を実践しています。
【参考文献】
- Yoshinaga M, et al. Probability of diagnosing long QT syndrome in children and adolescents according to the criteria of the HRS/EHRA/APHRS expert consensus statement. European heart journal. 2016;37:2490-7.
- 日本循環器学会/日本心臓病学会/日本小児循環器学会合同ガイドライン 2024 年改訂版 心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関するガイドライン
- Wilde AAM, et al. EHRA/HRS/APHRS/LAHRS Expert Consensus Statement on the state of genetic testing for cardiac diseases. J Arrhythm. 2022;38:491-553.
- Aiba T, et al. Clinical Impact of Genetic Testing for Long QT Syndrome - Evidence From a Nationwide LQTS Registry in Japan. Circ J. 2025. doi: 10.1253/circj.CJ-25-0105
- 日本循環器学会/日本小児循環器学会合同ガイドライン 2025 年JCS/JSPCCSガイドラインフォーカスアップデート版「学校心臓検診のガイドライン」
最終更新日:2025年04月18日